© Paesani Research Group. All rights reserved.
Publications 2012
57. Toward a universal water model: First principles simulations from the dimer to the liquid phase. V. Babin,
G.R. Medders, F. Paesani, J. Phys. Chem. Lett. 3, 3765 (2012). [link]
A full-dimensional model of water, HBB2-pol, derived entirely from “first-principles”, is introduced and
employed in computer simulations ranging from the dimer to the liquid. HBB2-pol provides excellent
agreement with the measured second and third virial coefficients and, by construction, reproduces the
dimer vibration–rotation–tunneling spectrum. The model also predicts the relative energy differences
between isomers of small water clusters within the accuracy of highly correlated electronic structure
methods. Importantly, when combined with simulation methods that explicitly include zero-point energy
and quantum thermal motion, HBB2-pol accurately describes both structural and dynamical properties
of the liquid phase.

56. Water structure, dynamics, and spectral signatures: Changes upon model cavity-ligand recognition. R. Baron,
P. Setny, F. Paesani, J. Phys. Chem. B 116, 13774 (2012). [link]
In this study, hydrophobic cavity-ligand association in a model system is characterized through
the analysis of the structure, dynamics, and corresponding spectral signatures of water at different
stages of the binding process. Molecular dynamics simulations reveal that the reorientation of the
water molecules around the ligand becomes faster as the receptor-ligand distance reduces, which
is correlated with the decrease in number of water-water hydrogen bonds within the ligand hydration
shells. The analysis of both linear and nonlinear infrared spectra allows direct insight into the evolution
of water structure and dynamics around the ligand. In particular, characteristic spectroscopic features
emerge at key stages of the binding process, which are related to changes in the hydrogen-bond
topology of water around the ligand. This study demonstrates that computer simulations and
vibrational spectroscopy could be integrated to facilitate the direct study of solvent effects in
biomolecular association.

55. The effects of electronic polarization on water adsorption in metal-organic frameworks: H2O in MIL-53(Cr).
J. Cirera, J.C. Sung, P. Howland, F. Paesani, J. Chem. Phys. 137, 054704 (2012). [link]
The effects of electronic polarization on the adsorption of water in the MIL-53(Cr) metal-organic framework
are investigated using molecular dynamics simulations. For this purpose a fully polarizable force field for
MIL-53(Cr) was developed which is compatible with the ab initio-based TTM3-F water model. The analysis
of the spatial distributions of the water molecules within the MIL-53(Cr) nanopores calculated as a function
of loading indicates that polarization effects play an important role in the formation of hydrogen bonds
between the water molecules and the hydroxyl groups of the framework. As a result, large qualitative
differences are found between the radial distribution functions calculated with non-polarizable and
polarizable force fields. The present analysis suggests that polarization effects can significantly impact
molecular adsorption in metal-organic frameworks under hydrated conditions.

54. Theoretical prediction of spin-crossover temperatures in ligand-driven light-induced spin change systems.
J. Cirera, F. Paesani, Inorg. Chem. 51, 8194 (2012). [link]
Spin-crossover compounds exhibit two alternative spin states with distinctive chemical and
physical properties, a particular feature that makes them promising materials for
nanotechnological applications. In this study, a theoretical/computational approach is described
for the calculation of the spin-crossover temperature (T1/2) for the trans-[Fe(styrylpyridine)4(NCX)2]
(X = S, Se, and BH3, styrylpyridine in the trans configuration) ligand driven light-induced spin
change (LD-LISC) complexes. In all cases, the calculations provide an accurate description of
both structural and electronic properties of the LD-LISC complexes and, importantly, predict
spin-crossover temperatures in good agreement with the corresponding experimental data.
Fundamental insights into the dependence of T1/2 on the nature of the axial ligands are
obtained from the direct analysis of the underlying electronic structure in terms of the relevant
molecular orbitals.

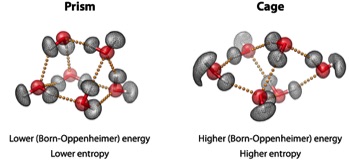
53. The water hexamer – cage, prism or both: Full dimensional quantum simulations say both. Y. Wang, V. Babin,
J.M. Bowman, F. Paesani, J. Am. Chem. Soc. 134, 11116 (2012). [link]
State-of-the-art quantum simulations on a full-dimensional ab initio potential energy surface are
used to characterize the properties of the water hexamer. The relative populations of the different
isomers are determined over a wide range of temperatures. While the prism isomer is identified
as the global minimum-energy structure, the quantum simulations, which explicitly include
zero-point energy and quantum thermal motion, predict that both the cage and prism isomers are
present at low temperature down to almost 0 K. This is largely consistent with the available
experimental data and, in particular, with very recent measurements of broadband rotational
spectra of the water hexamer recorded in supersonic expansions
52. Application of adaptive QM/MM methods to molecular dynamics simulations of aqueous systems. K. Park,
A.W. Götz, R.C.Walker, F. Paesani, J. Chem. Theory Comput. 8, 2868 (2012). [link]
The difference-based adaptive solvation quantum mechanics/molecular mechanics (adQM/MM) method
(J. Chem. Theory Comput. 2009, 5, 2212) as implemented in the Amber software was applied to the study
of several chemical processes in solution. The adQM/MM method is based on an efficient selection scheme
that enables quantum-mechanical treatment of the active region of a molecular system in solution taking
explicitly into account diffusion of solvent molecules between the QM and the MM regions. Specifically,
adQM/MM molecular dynamics simulations are carried out to characterize (1) the free energy profiles of
halide exchange SN2 reactions in water, (2) the hydration structure of the Cl- ion, and (3) the solvation
structure of the zwitterionic form of glycine in water. A comparison is made with the results obtained using
standard MM and QM/MM methods as well as with the available fully QM and experimental data. In all cases,
it is shown that the adaptive QM/MM simulations provide a physically realistic description of the system
of interest.

51. Water in metal-organic frameworks: Structure and diffusion of H2O in MIL-53(Cr) from quantum simulations.
F. Paesani, Mol. Simul. 38, 631 (2012). [link]
The structural and dynamical properties of water in the nanopores of MIL-53(Cr) are examined using
classical and quantum molecular dynamics simulations. The results indicate that, depending on the
number of molecules adsorbed per unit cell as well as on the shape of the nanopores, the explicit
inclusion of nuclear quantum effects can either enhance or inhibit the molecular mobility. At low loadings,
when MIL-53(Cr) exists in a narrow pore configuration, the translational and rotational motion of the water
molecules is largely suppressed. Importantly, it is found that nuclear quantum effects make the reorientation
of the water molecules within the narrow nanopores slower than in the classical limit. This is attributed to
the quantum delocalisation that effectively increases the molecular volume and, consequently, reduces the
free space available in the nanopores. As the number of molecules per unit cell increases and the
nanopores start opening, the dynamics of quantum water becomes faster again.

50. Molecular level characterization of the breathing behavior of the jungle-gym-type DMOF-1 metal-organic
framework. J.S. Grosch, F. Paesani, J. Am. Chem. Soc. 134, 4207 (2012). [link]
Fundamental insights into the molecular mechanisms that determine the breathing behavior of
the jungle-gym-type DMOF-1 metal–organic framework upon adsorption of benzene andisopropyl
alcohol are gained from computer simulations. In all cases, good agreement is obtained between
the calculated and experimental structural parameters. In the case of benzene adsorption, DMOF-1
is predicted to exist in a narrow pore configuration at high loadings and low temperature. A structural
transition into a large pore configuration is then observed as the temperature increases and/or the
loading decreases, which is directly related to the spatial distribution and molecular interactions
of the benzene molecules within the pores. The isopropyl alcohol adsorption simulations indicate
that DMOF-1 undergoes two distinct structural transitions (from large pore to narrow pore and then
back to large pore) as the number of adsorbed molecules increases, which is related to the
formation of hydrogen bonds between the isopropyl molecules and the framework.

49. A refined MS-EVB model for proton transport in aqueous environments. K. Park, W. Lin, F. Paesani,
J. Phys. Chem. B 116, 443 (2012). [link]
In order to improve the description of proton mobility in aqueous environments, a revised multistate
empirical valence bond model (aMS-EVB3) is developed. The new aMS-EVB3 model is built upon
an anharmonic water force field (aSPC/Fw) in which the OH bond potential is described through
a quartic approximation to a Morse potential. First, it is shown that the aSPC/Fw anharmonic water
model provides an accurate description of water at ambient conditions and reproduces the available
experimental data for several structural, thermodynamic, and dynamical properties. Second, it is
shown that, when applied to the study of proton solvation and transport in bulk water, the new
aMS-EVB3 model accurately describes the solvation structure around the excess proton.
Importantly, the new aMS-EVB3 model predicts a arger proton diffusion coefficient than previous
models, which largely improves the agreement with the available experimental data.